Quickstart¶
Overview¶
SevenBridges CWL package provides python bindings for Common Workflow Language v1.0. It is intended for developers who want to use python code to generate CWL documents. If creating a document through the GUI is preferable, then look at the Rabix Composer. This library also have integration with sevenbridges-python so applications can be easily deployed on a Seven Bridges platform.
Tool creation¶
Docker image preparation¶
Your code will run in a docker image that you have prepared before defining and
running a tool. All libraries needed for your function to run must be
installed on the image. The only additional requirement for sevenbridges-cwl
is to have dill package and bzip2 installed on the image - so when you
prepare your image pip install dill and apt-get install bzip2 on it.
Wrapping binaries¶
One way of creating a tool is by using CommandLineTool class which is
useful for wrapping binaries:
from sbg import cwl
t = cwl.CommandLineTool(
base_command=['echo', 'HelloWorld'],
stdout='_stdout_',
requirements=[cwl.Docker(docker_pull='ubuntu:16.04')]
)
t.add_output(cwl.File(glob='_stdout_', required=True), id='out')
Example above illustrates echo HelloWorld > _stdout_ command. Object t is
an instance of CommandLineTool class which has a number of useful builtin
methods described here.
Generated tool can be easily run on a Seven Bridges platform using
Session object:
session = cwl.Session(profile='<your_profile>')
session.run('<your_project>', t)
If inspecting raw CWL documents is preferable use cwl.tool context manager:
from sbg import cwl
with cwl.tool('hello_world.cwl', 'w') as t:
t.base_command = ['echo', 'HelloWorld'] # echo 'HelloWorld' on stdout
t.add_requirement(cwl.Docker(docker_pull='ubuntu:16.04'))
t.stdout = '_stdout_' # redirect all stdout to this '_stdout_' file
t.add_output(cwl.File(glob='_stdout_', required=True), id='out')
First parameter to the tool function is a file path for CWL document.
Second parameter is file access which can be either one of:
w- for writingr- for readingrw- for editing
After running code block above, hello_world.cwl file is created and dumped
into the current working directory with contents:
baseCommand:
- echo
- HelloWorld
class: CommandLineTool
cwlVersion: v1.0
inputs: []
outputs:
- id: out
outputBinding:
glob: _stdout_
type: File
requirements:
- class: DockerRequirement
dockerPull: ubuntu:16.04
stdout: _stdout_
Wrapping python code¶
Tool can be created using @to_tool decorator only by annotating python
function. Annotated functions are functions with defined types for inputs
and outputs, which is illustrated in the example below.
import pysam
from sbg import cwl
@cwl.to_tool(
docker='images.sbgenomics.com/filip_tubic/ubuntu1604pysam',
outputs=dict(out=cwl.File(glob='gc_content.txt'))
)
def gc_content(bam_file: cwl.File(secondary_files=['.bai']),
bed_file: cwl.File()):
"""Calculates GC content."""
bam_file = bam_file['path']
bed_file = bed_file['path']
bam = pysam.AlignmentFile(bam_file, 'rb')
with open('gc_content.txt', 'w') as out:
with open(bed_file) as bf:
for line in bf:
line_parts = line.strip().split()
chr = line_parts[0]
start = int(line_parts[1])
end = int(line_parts[2])
read_data = bam.fetch(chr, start, end)
total_bases = 0
gc_bases = 0
for read in read_data:
seq = read.query_sequence
total_bases += len(seq)
gc_bases += len([x for x in seq if x == 'C' or x == 'G'])
if total_bases == 0:
gc_percent = 'No Reads'
else:
gc_percent = '{0:.2f}%'.format(
float(gc_bases) / total_bases * 100
)
out.write('{0}\t{1}\n'.format(line.strip(), gc_percent))
Function gc_content will accept .bam and .bed files and calculates
GC content for each interval defined in bed file. Corresponding output
will be dumped into a gc_content.txt file. After running code above,
command line tool will be created with already set inputs
(bam_file, bed_file) and output (out). In order to run this function we
use Session.
session = cwl.Session(profile='<your_profile>')
project = '<your_project>'
files = list(session.api.files.query(
project=project,
names=['<bam_file>', '<bed_file>']
))
session.run(project, gc_content(), inputs=dict(
bam_file=files[0],
bed_file=files[1]
))
NOTE After generating tool fromgc_contentfunction, base command will be set topython{major}.{minor} gc_content.pywhere{major}.{minor}is python version that is used for calling code block above. So if you’re using python 3.6 locally you need to have python 3.6 installed in your docker image.
Input/Output types are translated into CWL concrete types by following rules:
cwl.Int()is converted into cwl integercwl.String()is converted into cwl stringcwl.Float()is converted into cwl floatcwl.Bool()is converted into cwl booleancwl.File()is converted into cwl filecwl.Dir()is converted into cwl directorycwl.Record(k1=cwl.String(), k2=cwl.Int())is conveted into cwl record withstringandintas field types namedk1andk2respectivelycwl.Union()is converted intoUniontype (can be either one of specified types, eg:cwl.Union([cwl.Int(), cwl.String()])- int or string)cwl.Enum()is converted into cwl enumcwl.Array(<t>)is converted into cwl array of typet(eg.cwl.Array(cwl.Int())- list of ints)
Complete documentation of @to_tool decorator is located
here.
Wrapping bash code¶
Tools can be generated from existing bash scripts using cwl.from_bash
function.
from sbg import cwl
t = cwl.from_bash(
label='Example of bash tool',
inputs=dict(
STR=cwl.String(),
),
outputs=dict(
out=cwl.File(glob='stdout')
),
script=r'''echo $STR''',
stdout='stdout',
docker='images.sbgenomics.com/filip_tubic/ubuntu1604bzip'
)
Workflow creation¶
Workflow can be easily created from existing tool objects. One way of creating
workflow can be done using with workflow(...) statement.
from sbg import cwl
# First node
@cwl.to_tool(
inputs=dict(x=cwl.String()),
outputs=dict(out=cwl.Float(required=True)),
docker='images.sbgenomics.com/filip_tubic/ubuntu1604py'
)
def to_float(x):
return dict(out=float(x))
# Second node
@cwl.to_tool(
inputs=dict(x=cwl.Float(), n=cwl.Int()),
outputs=dict(out=cwl.Float()),
docker='images.sbgenomics.com/filip_tubic/ubuntu1604py'
)
def times_n(x, n=10):
return dict(out=x * n)
with cwl.workflow('wf.cwl', 'w') as wf:
# create tools
t1 = to_float()
t2 = times_n()
# steps
wf.add_step(t1, expose=['x'])
wf.add_step(t2, expose=['n', 'out'])
# add connections
wf.add_connection('{}.out'.format(t1.id), '{}.x'.format(t2.id))
Object wf is an instance of Workflow class which documentation can be found
here.
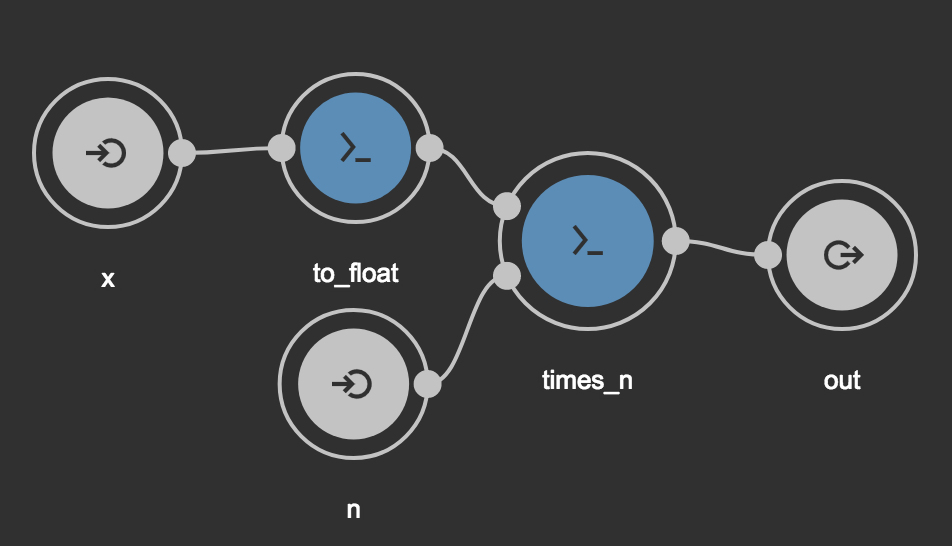
Running code block above will generate wf.cwl in the current working
directory. Using Rabix Composer generated
file can be easily visualized as a graph. By pasting contents of wf.cwl in
the Code section in Rabix composer, following graph
will be displayed in Visual Editor section. Like in examples before, we use
Session to run workflow.
{kind=link}
from sbg import cwl
# First node
@cwl.to_tool(
inputs=dict(x=cwl.String()),
outputs=dict(out=cwl.Float(required=True)),
docker='images.sbgenomics.com/filip_tubic/ubuntu1604py'
)
def to_float(x):
return dict(out=float(x))
# Second node
@cwl.to_tool(
inputs=dict(x=cwl.Float(), n=cwl.Int()),
outputs=dict(out=cwl.Float()),
docker='images.sbgenomics.com/filip_tubic/ubuntu1604py'
)
def times_n(x, n=10):
return dict(out=x * n)
wf = cwl.Workflow()
# create tools
t1 = to_float()
t2 = times_n()
# steps
wf.add_step(t1, expose=['x'])
wf.add_step(t2, expose=['n', 'out'])
# add connections
wf.add_connection('{}.out'.format(t1.id), '{}.x'.format(t2.id))
# Session on a SBG platform
session = cwl.Session(profile='<your_profile>')
session.run('<your_project>', wf, inputs={'x': '10.2', 'n': 10})
Loading existing documents¶
Existing CWL documents can be loaded from a file using load function,
docs.
from sbg import cwl
t = cwl.CommandLineTool(
base_command=['echo', 'Hello'],
).dump('dummy.cwl')
x = cwl.load('dummy.cwl')
print(' '.join(x.base_command)) # prints 'echo Hello'
assert isinstance(x, cwl.CommandLineTool)